|
|
We have developed an algorithm for rapid computation of the smooth molecular surface. Our
algorithm is analytical, easily parallelizable, and generates triangulated molecular surface.
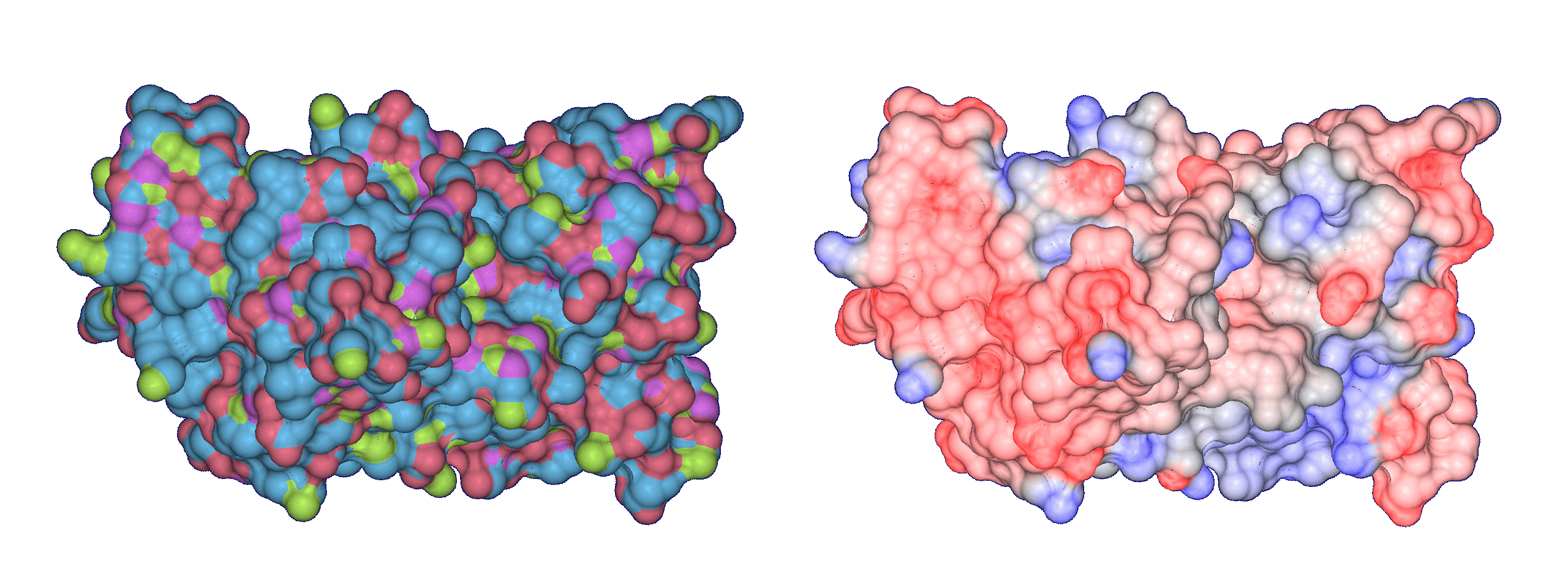
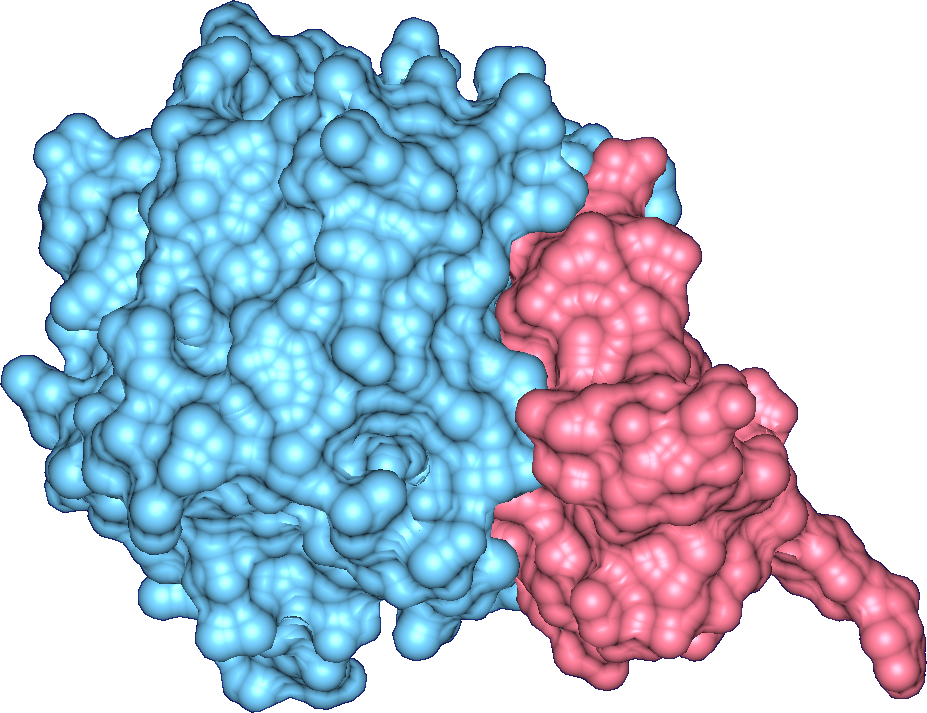
One of the important factors that influences the position and orientation of the protein with
respect to the substrate in protein-substrate docking is the geometric fit or surface
complementarity. Traditionally, the interface has been studied by using a clipping plane that
is moved along the z-axis in the screen space. This does not readily convey the three-dimensional
structure of the interface to the biochemist. We have defined the molecular interface surfaces
between two molecular units, as a family of surfaces parametrized by the probe-radius
α and interface radius β.
The molecular interface surfaces are derived from approximations to the power-diagrams over the
participating molecular units. Molecular surfaces provide biochemists with a powerful tool to study
surface complementarity and to efficiently characterize the interactions during a protein-substrate
docking.
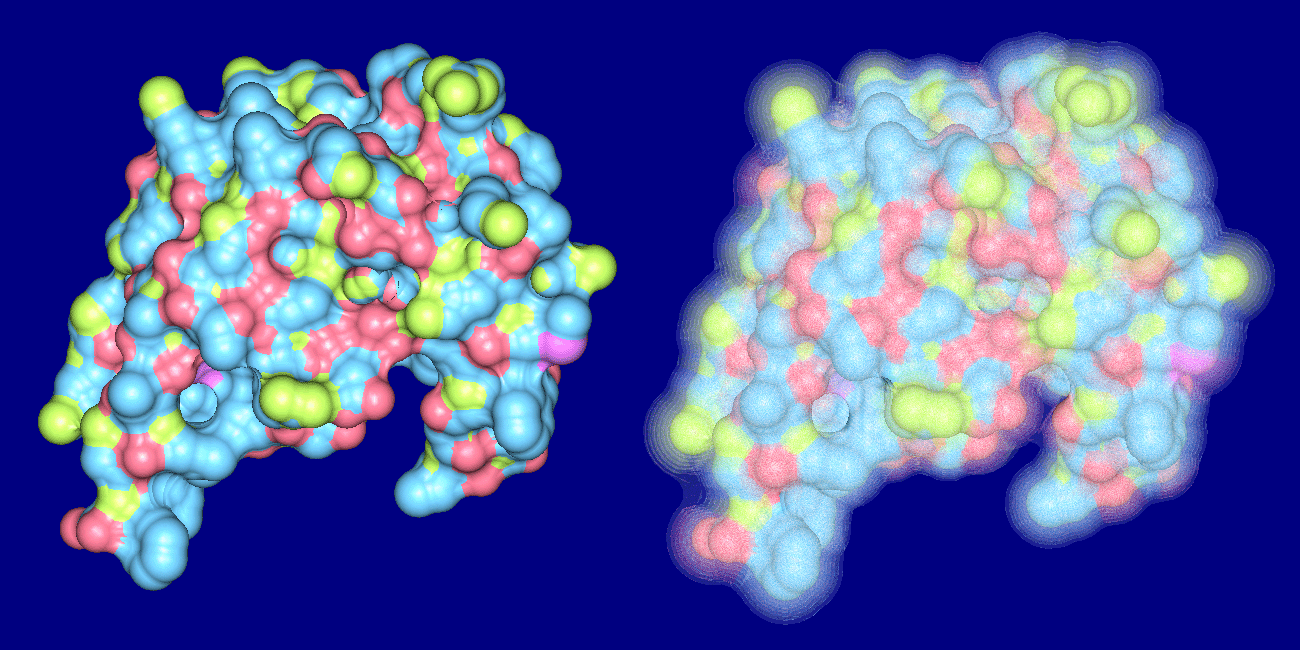
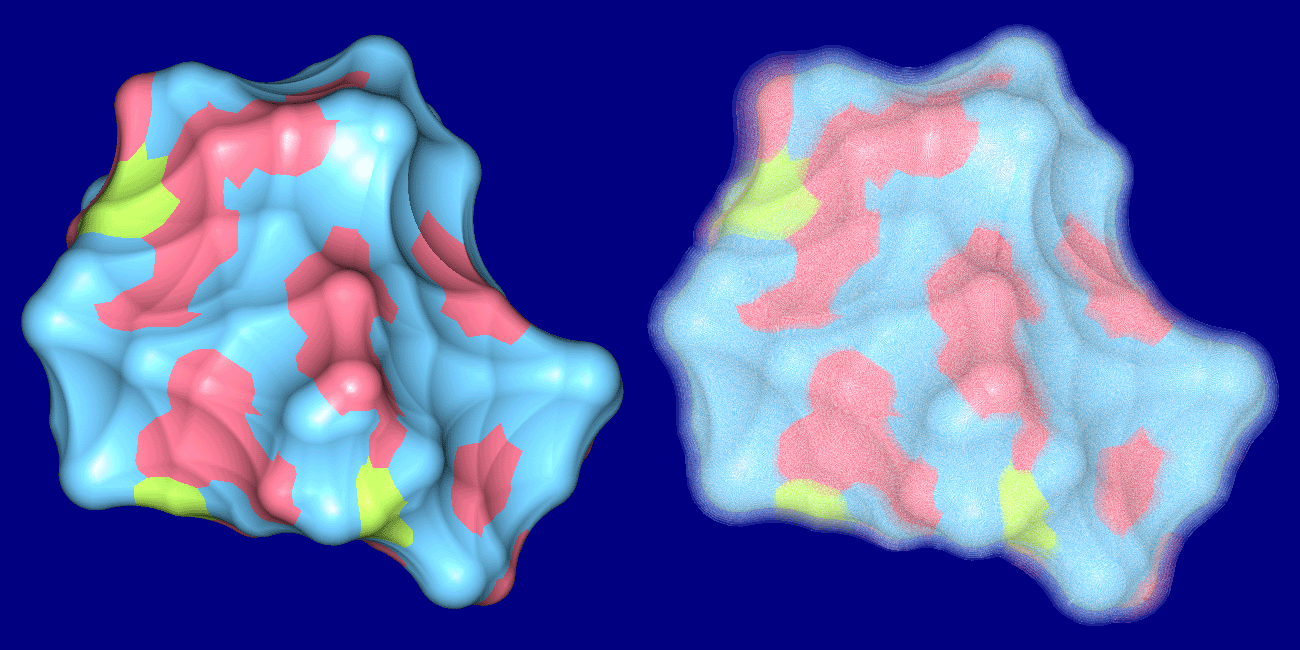
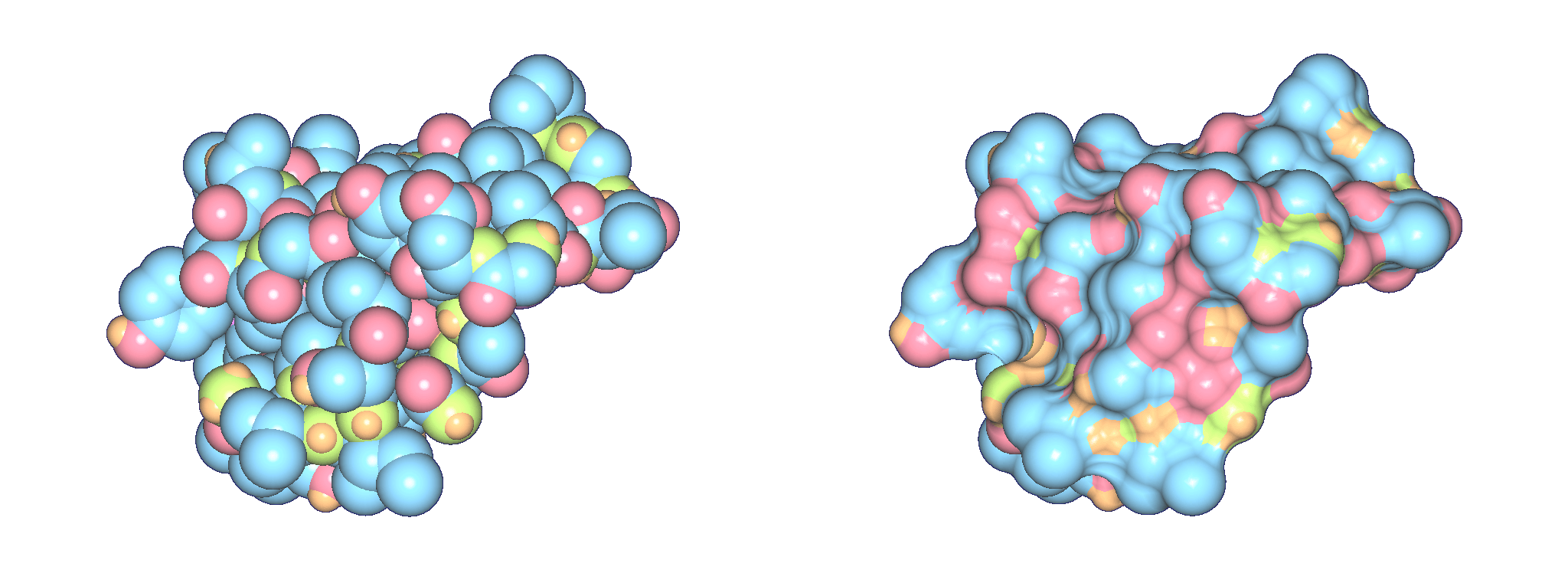
The top images show Crambin and its molecular surface (probe radius 1.4 Angstroms) and the bottom
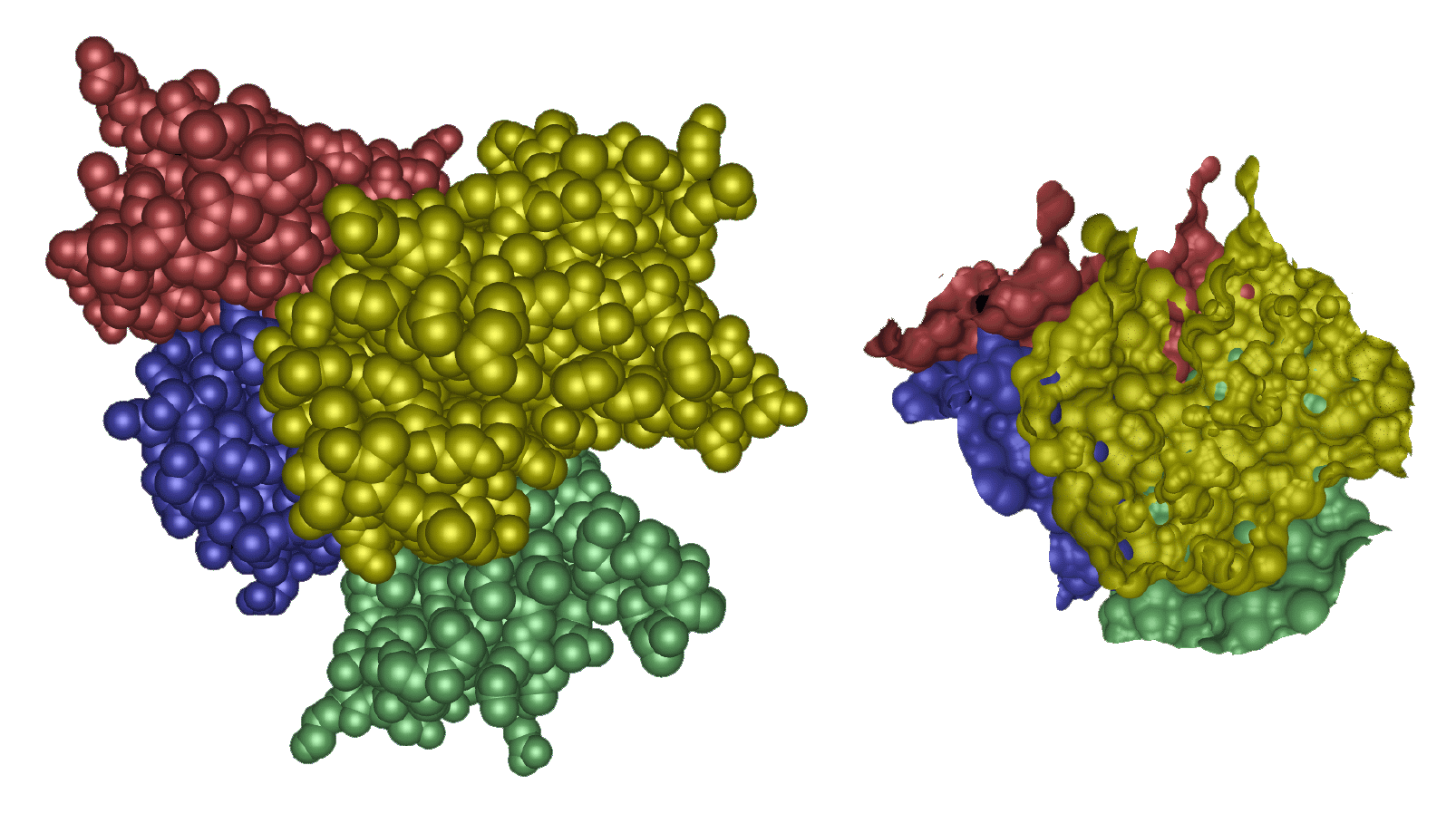
images show Trasthyretin domains and their molecular interface surface
(α = 1.0 Angstrom, β = 2.4 Angstroms).
-
Defining, Computing, and Visualizing Molecular Interfaces
A. Varshney, F. P. Brooks, Jr., D. C. Richardson, W. V. Wright, and D. Manocha
Proceedings of the IEEE Visualization '95
Oct 29 - Nov 3, 1995, Atlanta, GA, pp 36 - 43
-
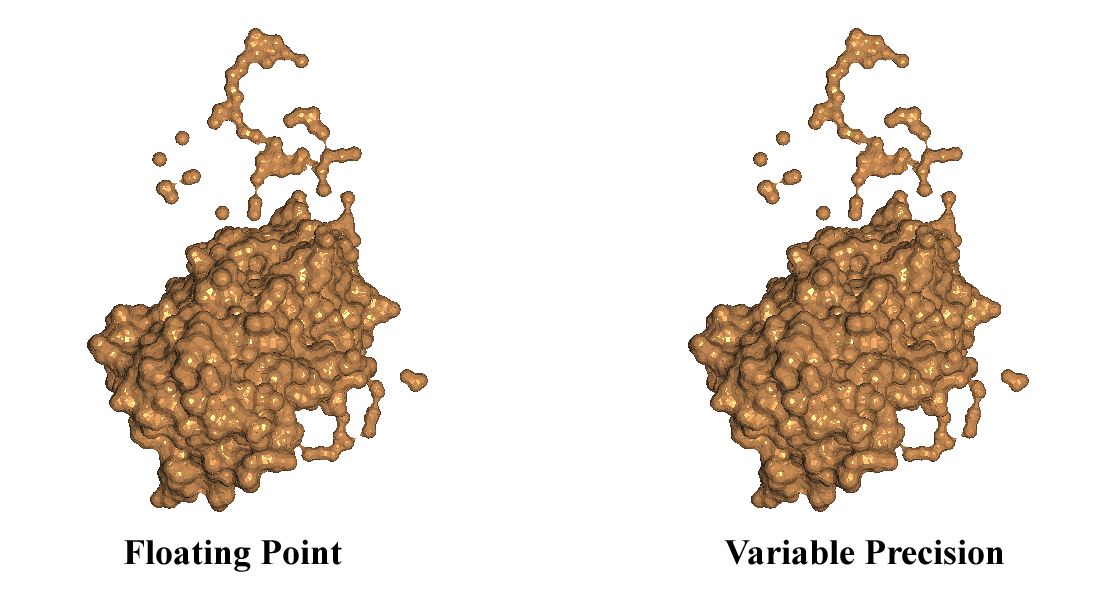
Linearly Scalable Computation of Smooth Molecular Surfaces
A. Varshney, F. P. Brooks, Jr., and W. V. Wright
IEEE Computer Graphics and Applications
Sept 1994, Vol. 14, No. 5, pp 19 - 25
-
Interactive Visualization of Weighted Three-dimensional Alpha Hulls
A. Varshney, F. P. Brooks, Jr., and W. V. Wright
Third Annual Video Review of Computational Geometry in Proceedings of the
Tenth Annual Symposium on Computational Geometry
Stony Brook, NY, June 6 - 8, 1994, pp 395 - 396
-
Fast Analytical Computation of Richards's Smooth Molecular Surface
A. Varshney and F. P. Brooks, Jr.
Proceedings of the IEEE Visualization '93
Oct 25 - 29, 1993, San Jose, CA, pp 300 - 307.