
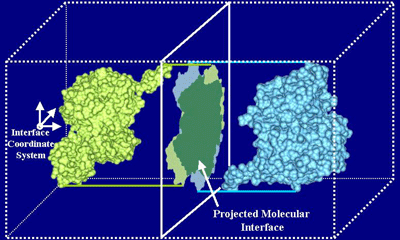
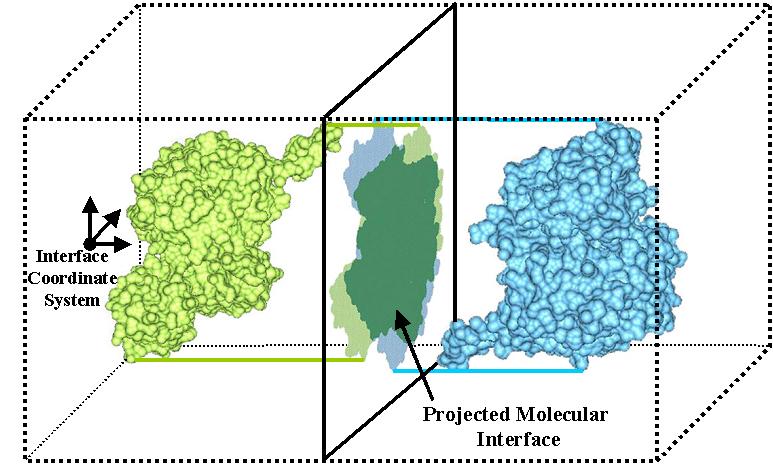
Protein docking is a Grand Challenge problem that is crucial to our understanding of biochemical processes. Several protein docking algorithms use shape complementarity as the primary criterion for evaluating the docking candidates. The intermolecular volume and area between docked molecules is useful as a measure of the shape complementarity. In this research we discuss an algorithm for interactively computing intermolecular negative volume and the area of docking site using graphics hardware. Our method leverages the recent advances in the 3D graphics hardware to achieve interactive rates of performance. Using this method scientists can interactively study various possible docking conformations and visualize the quality of the steric fit.

Several drug development processes have so far begun with large-scale random screening of candidate inhibitors. These initial discoveries are improved through well-defined approaches to find new drugs. As molecular structure determination techniques and computational methods progress, protein docking methods using structure-based molecular complementarity have become an important substitute for random screening in the drug design process.
Among many factors involved in protein-protein interactions such as electrostatics, hydrophobicity, and hydrogen bonding, shape complementarity is of major importance for protein docking. Purely geometric approach can restrict the time-consuming calculations of interaction energy to be performed only for those cases that have a good geometric fit. Geometric methods can also be used as foundations for more complete approaches considering chemical and energetic characteristics. A complete search of all possible geometric fits of two flexible molecules takes too much time because of the extremely large degrees of freedom. Therefore, molecules have been often assumed as rigid bodies. Even with the rigid body assumption, finding accurate shape complementarities remains a challenging problem. Most existing methods provide a list of candidates sorted by complementarity criteria and the final decision by human is needed. Therefore, an interactive tool for visualizing the shape complementarity would be useful.
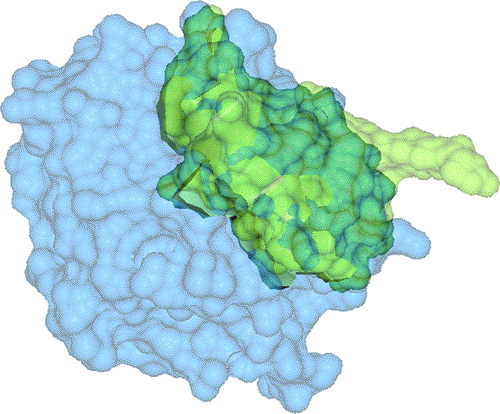

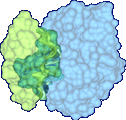
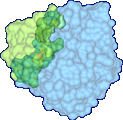
Correct Fit:
Area/Vol = 1.45Incorrect Fit:
Area/Vol = 0.83The main contributions of this research are:
- Our approach computes the intermolecular volume as well as the interface area accurately to any desired level of precision in linear time. The ratio of the area of the interface and intermolecular volume can be a good criterion for characterizing the goodness-of-fit for protein docking as well as rational drug design.
- We provide an interactive tool for visualizing the intermolecular negative volume to assist protein docking.
- We use the graphics hardware to accelerate the computation of the intermolecular negative volume for visualizing it at interactive rates.
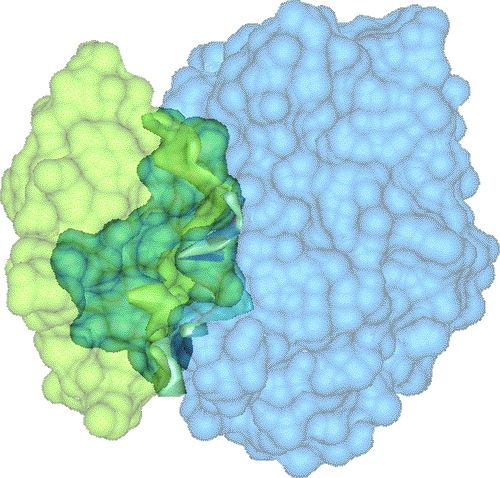
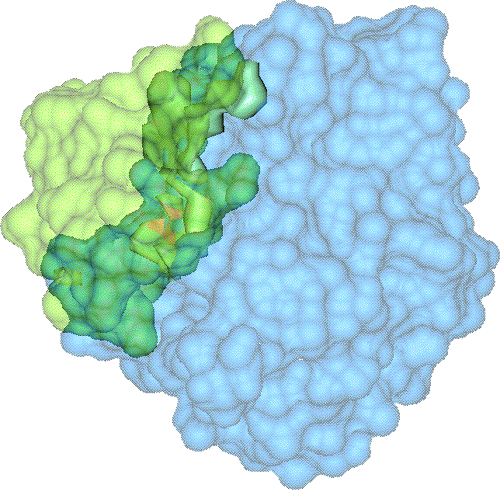
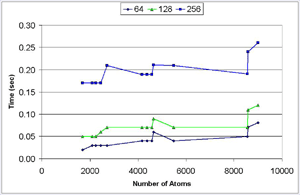
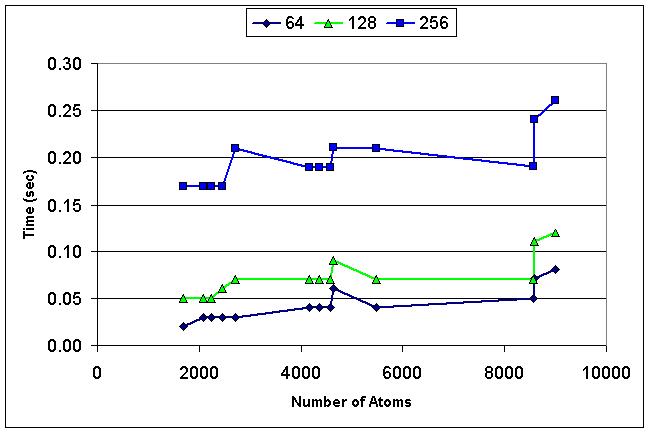
We have developed a 3D visualization tool for visualizing the intermolecular negative volume interactively, which can be used as a complementary tool to existing drug-design systems. Users can manipulate the molecules together or separately while the intermolecular negative volume is computed and rendered for every frame at interactive rates. Our system also provides various visualization options. The intermolecular negative volume can be visualized alone, or together with molecules as the images in this research show. The molecules can be visualized in solid surfaces or translucent surfaces. We have computed the intermolecular volumes, the interface area contributions from the two molecules, and the ratio of the average interface area to the intermolecular volume for a number of naturally occurring molecular complexes. We have computed the intermolecular volume at various resolutions: 64^3, 128^3, and 256^3. The computing time of our algorithm is linearly related to the number of atoms and runs at interactive rates as shown in the left figure. We have obtained these results on a Dell Precision Workstation with 1.5 GHz Pentium 4, 1 GB RAM, and a nVidia GeForce FX 5900 graphics card.



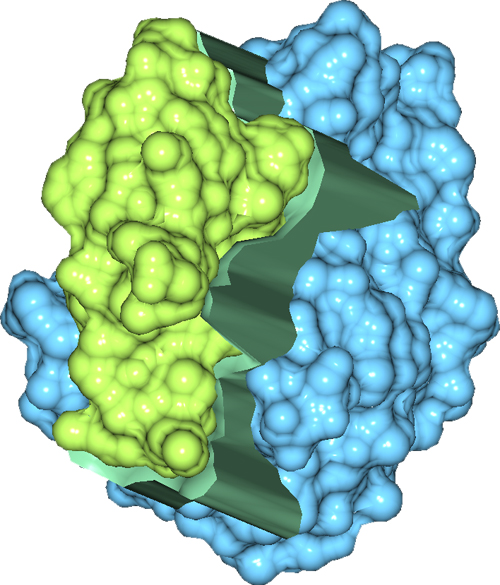
Our algorithm for computing intermolecular negative volume and the area of the molecular interfaces can be used for computing criteria for shape complementarity. Our 3D visualization tool for visualizing intermolecular negative volume interactively can be used as a complementary tool for existing protein docking and rational drug design systems. The visualization tool can be used to select the best fit among the candidates, and to improve the result by moving the molecules interactively to produce a better fit. Currently, we have only considered the geometric shape complementarity as a way of characterizing the goodness-of-fit between two molecules. Shape complementarity is an important criterion, but not the only one. It would be very helpful to include other criteria such as electrostatics, hydrophobicity, hydrogen bonding in developing a comprehensive metric for characterizing good molecular interfaces that can be used in protein docking and rational drug design applications.


- C. H. Lee and A. Varshney. Computing and Displaying Intermolecular Negative Volume for Docking. Scientific Visualization: Extracting Information and Knowledge from Scientific Datasets, Editors: G.-P. Bonneau, T. Ertl, G. M. Nielson, Springer-Verlag.
This work has been supported in part by the NSF grants: IIS 00-81847, CCF 04-29753, and CNS 04-03313. This material is based upon work supported by the National Science Foundation under grants IIS 00-81847, CCF 04-29753, and CNS 04-03313. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation.

Web Accessibility
